
Legal Action for Possible Philips CPAP Lung Injuries and Cancer Connections
If Philips has issued a recall on certain of their mechanical ventilators due to potential health concerns, including cancer. “The FDA has categorized this recall as a Class 1 recall, the most serious type of recall. Use of these devices may cause serious injuries or death.” If you used a Philips mechanical ventilator between November 5th 2005 and April 23, 2021, you may be entitled to financial compensation through a lawsuit.
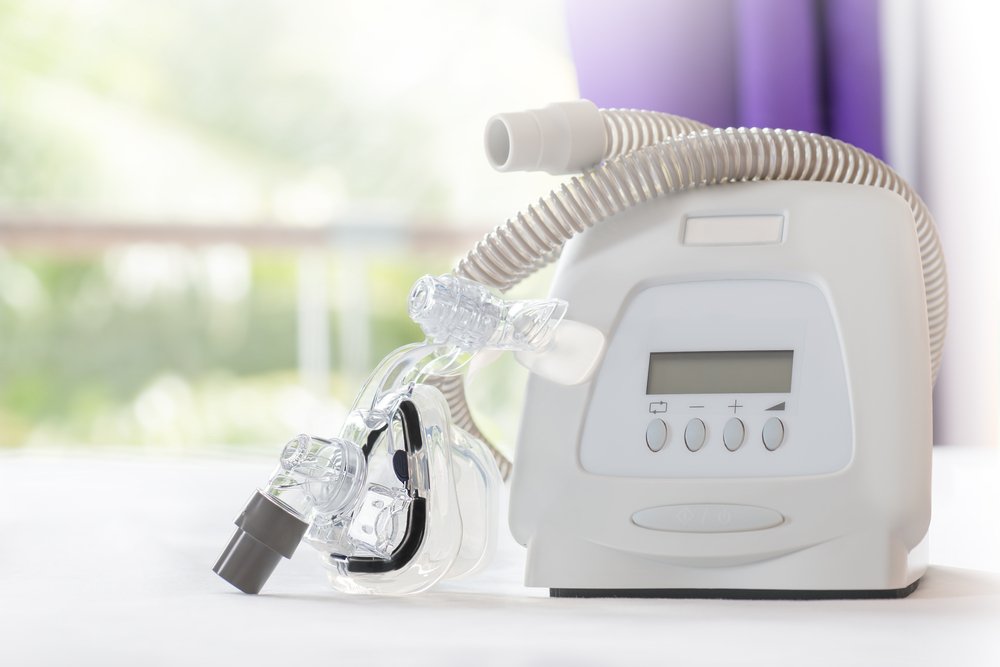
What are CPAP & BiPAP Machines?
CPAP and BiPAP Machines are used to treat certain types of Sleep Apnea. Sleep Apnea is a serious sleep disorder in which breathing is momentarily interrupted and may stop for a brief period of time. Many patients who suffer from Sleep Apnea may be recommended the use of a CPAP (Continuous Positive Airway Pressure) machine. CPAP therapy works to maintain open, obstruction-free airway through the flow of pressurized air, to keep the airway open. The machine delivers airflow at a fixed pressurized setting through a tube, which is attached to the machine. The demand for CPAP Machines and Ventilators has skyrocketed recently, in part due to Covid-19. CPAP sales generated roughly $6 billion in revenue for Philips in 2020.
Information About the Recall
Philips publicly announced a broad safety recall of several CPAP, BiPAP and Ventilators, along with the DreamStation series, on June 14, 2021, due to Potential Health Risks related to the PE-PUR Sound Abatement Foam. It is believed that this Polyester-based polyurethane foam may release particles or off-gas certain chemicals that can cause a chronic inflammatory response, lack of oxygen, and other toxic reactions.
Polyester polyurethane may be quickly broken down by heat, sunshine, moisture, microbial, fungal attack, and even oxygen, which is an issue with PE-PUR foam. If your equipment, for example, was exposed to excessive humidity or temperatures, you are more vulnerable to this recall.
How Many Devices Are Affected?
Prior to the FDA officially recalling Zantac and Ranitidine products, numerous brands vReports show that there are roughly 15 million machines that were sold between 2005 and April 2021, which could contain the defective foam. The majority of these recalled machines belong to the “DreamStation” line of the CPAP machines, which was launched in October 2015.
If you have used a CPAP machine and have been injuried, you may be eligible for significant compensation. Bighorn Law can help you today.
Injuries
Some of the symptoms associated with CPAP Machine Foam Deterioration are:
- American Health Packaging
- Amneal Pharmaceuticals
- Aurobindo Pharma USA
- Apotex Corp.
- Respiratory Tract Irritation
- Persistent Cough
- Chest Pressure
- Interstitial Lung Disease
- Pulmonary Fibrosis
- Sarcoidosis
- Pneumoconiosis
- Auto-Immune Disorders
- Chronic Bronchitis
- Kidney Disease
- Liver Disease
- Cancer, Lymphoma, Leukemia or Tumors
- Heart Failure
Philips Recalled Devices
A-Series BiPAP A30® and Hybrid A30®
A-Series BiPAP A40®
A-Series BiPAP V30 Auto®
C-Series ASV®
C-Series S/T® and AVAPS®
Dorma 400®
Dorma 500®
DreamStation®
DreamStation ASV®
DreamStation Go®
DreamStation ST® and AVAPS®
E30®
Garbin Plus®, Aeris®, LifeVent®
OmniLab Advanced+®
REMstar SE Auto®
SystemOne ASV4®
SystemOne (Q-Series)®
Trilogy 100®
Trilogy 200®
Bighorn Law knows the CPAP lawsuit situation and can get you compensated if you have a connection between the device and injuries.
What Can I Get In a CPAP Lawsuit?
You can receive a settlement that takes into account past and future medical bills, in addition to lost earnings. Noneconomic losses can also be recovered for the emotional pain and suffering that you have endured due to the situation.
What is a Medical Device?
A medical device is an instrument, implant, or other article that is intended for use to cure, prevent, or treat a disease and is not otherwise generally considered a drug. The FDA classifies every medical device based on its level of risk to a patient:
- Class I medical devices are considered the lowest risk and include such things as elastic bandages and examination gloves.
- Class II medical devices are considered a moderate risk of harm to consumers, and this includes such things as CPAP machines, powered wheelchairs, and some pregnancy test kits.
- Class III medical devices are those that have the potential for a high risk of injury to consumers when they are implanted and otherwise pose significant danger. They include such things as breast implants, implantable pacemakers, and some hip implants.
Are Medical Devices Approved By The FDA?
The FDA is the U.S. Food and Drug Administration. It is the federal government agency that is tasked with protecting the public health by overseeing the sale of food and drugs in the United States. Their mission is also to stimulate and promote innovation in the sciences and to provide consumers and patients with science based information on which they can make decisions.
Before a medical device, such as a CPAP machine, can be marketed in the United States, it must go through a clearance process. There are actually two types of clearance processes governed by the FDA:
- The pre-market approval process; and
- The pre-market notification process
The difference between these two processes is very important.
Class I and II devices can go through the pre-market notification process in which the FDA determines that the device is substantially equivalent to another device that was previously allowed to be marketed in the United States. The more rigorous pre-market approval process is reserved for Class III devices with much higher risk of injury and requires that the party seeking approval to market the product prove that it is safe and effective for use.
What Does FDA Approval Mean To A Patient
As a patient, you want to know everything you can before making a decision to undergo a procedure in which a medical device is implanted in your body. One of the things you should want to know is whether the device was cleared or approved by the FDA.
Then, if the device were cleared through the pre-market notification process, a patient needs to understand that the level of testing the device underwent prior to it being marketed in the United States is not as high as the level of testing required for the pre-market approval process. What’s more, even if the device went through the more rigorous pre-market approval process, this does not necessarily mean that the device is safe.
Finally, if you have been harmed by a defective medical device, the fact that it was approved by the FDA does not mean that you cannot hold the manufacturer liable to compensate you for injuries and losses you suffered as a result of using their defective product. In Nevada, the manufacturer, distributor, and any other entity in a defective product’s line of distribution can be held strictly liable for injuries caused to those who use the product the way it was intended to be used.
Bighorn Law will fight for you in your CPAP lawsuit in Las Vegas. Talk to someone today about your situation by calling (702) 333-1111.